Developing non-precious metal catalysts often requires exploration of new mechanistic paradigms.
One theme emerging from
our bioinorganic model studies
is the pervasive role of M 𝛿+···M𝛿- pairs in cooperative bond activation at multimetallic clusters.
Partly inspired by that observation, we have pursued a program developing heterobinuclear transition metal pairs for cooperative catalysis.
Transition metal Lewis pairs.
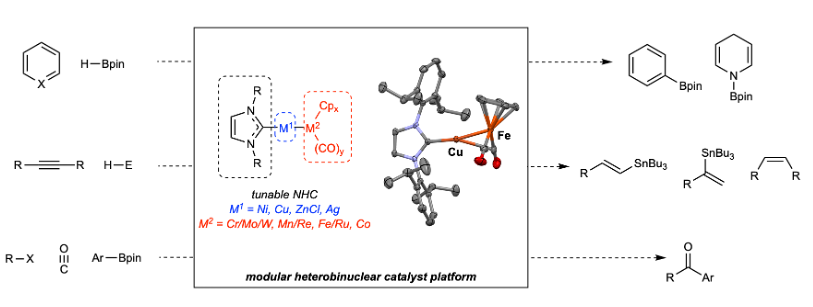
Since 2013, our group has studied heterobinuclear catalysts of the type (NHC)M–[MCO], where NHC is a tunable N-heterocyclic carbene; M is usually Cu but also Ni, ZnCl, or Ag; and [M CO]- is a metal carbonylate such as [FeCp(CO)2]-, [Mn(CO)2 ]-, [MoCp(CO)3]-, etc.
These complexes feature M ← MCO bonds that are predominantly dative in character and are believed to dissociate reversibly at ambient conditions to provide access to equilibrium concentrations of [(NHC)M]+/[MCO]- Lewis acid/base pairs.
These transition metal frustrated Lewis pairs (FLPs) were successfully used to catalyze various organic transformations that rely on cooperative activation of H–H, H–B, or H–Sn bonds and/or cooperative formation of C–H or C–C bonds.
Examples include C–H borylation, alkyne semi-hydrogenation, CO2 hydroboration, pyridine hydroboration, alkyne hydrostannylation, and carbonylative Suzuki-Miyaura coupling.
In addition to developing these catalytic transformations, we have expended significant efforts to study the cooperative bond activation mechanisms using a combination of experimental and computational methods, and these systems have also inspired experimental and computational studies from other groups.
Metalloradical pairs.
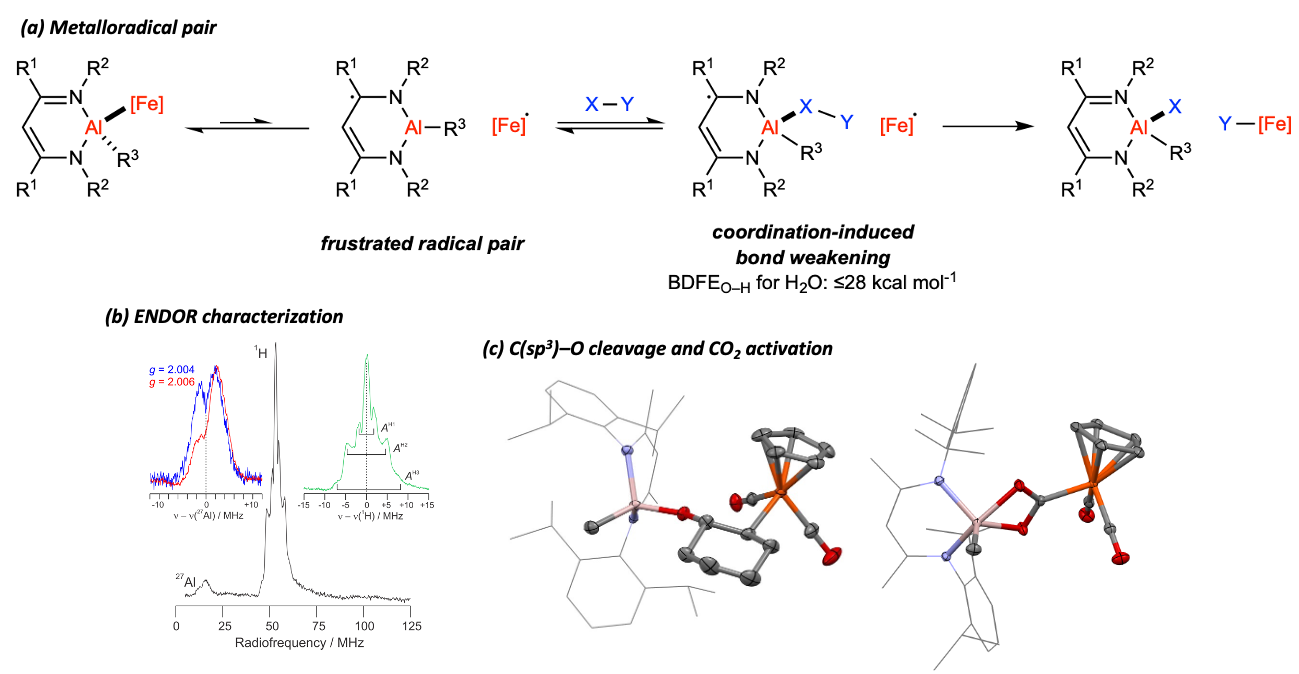
Recently, we became motivated to pursue cooperative chemistry of Al and Fe, which are earth’s two most abundant metals. We have found a Al–Fe complex, LdippAl(Me)FeCp(CO)2, developed in our lab shows diverse reactivity towards O- and N-substrates, including activation of π-systems in CO2 and pyridine and cleavage of robust 𝜎-bonds such as the C(sp3)–O linkages in cyclic ethers.
Mechanistic studies indicate that the reactivity profile is an example of frustrated radical pair (FRP) chemistry, which is a frontier area of catalysis research.
In other words, the Al–Fe bond undergoes reversible homolysis at ambient conditions, producing equilibrium concentrations of the [LdippAlMe]·/[FeCp(CO)2]· FRP.
Bond activation then occurs via dramatic coordination-induced bond weakening (CIBW) of substrates at the Al(III) center of redox non-innocent [LdippAlMe]·.
We estimate that the O–H bond of H2O is weakened from 113 kcal mol-1 to ≤ 28 kcal mol-1 by CIBW in this case.
Radical intermediates were characterized by multi-frequency EPR and ENDOR spectroscopy.
Our long-term goal with this and related Al-containing FRPs is to develop catalytic C(sp3)–O functionalization reactions.
To do this, we have embarked on a catalyst design campaign that is driven by computational modeling and data science.
Continuation of such analyses will lead to “intelligent design” of second-generation Al–Fe catalysts for targeted transformations.
